The FDA has regularly engaged with congressional and industry stakeholders to revise its general approach of enforcement discretion on LDTs since 2010. This collaboration allows the FDA to align with the evolution of LDTs and to ensure their safety and effectiveness. In October 2023, the FDA issued a proposed rule for this purpose, with the rulemaking finalized in April 2024 and the final rule published on May 6, 2024.
The FDAs Final Rule on LDTs
In this article, we provide an overview of the impacts of this final rule, which phases out the FDA’s general enforcement discretion approach for LDTs.
Are you a laboratory that manufactures LDTs? MedEnvoy can assist you in navigating these complex requirements as a result of this rule change. Click here, for more information.
What is an FDA LDT?
The FDA has traditionally considered LDTs to be in vitro diagnostics (IVDs) intended for clinical use and designed, manufactured, and used within a single laboratory certified under the Clinical Laboratory Improvement Amendments (CLIA) OF 1988 and meets the regulatory requirements under CLIA to perform high complexity testing (e.g. pertinent laboratory requirements established under 42 CFR Part 493).
The phaseout policy under the final rule applies to “IVDs offered as LDTs” which are IVDs manufactured and offered as LDTs by laboratories that are certified under CLIA and meet the regulatory requirements under CLIA to perform high complexity testing, and used within such laboratories, even if those IVDs do not fall within the FDA’s traditional understanding of a LTD because they are not designed, manufactured, and used within a single laboratory.
The FDA is also amending the definition of “in vitro diagnostic products” in its regulations (e.g. 21 CFR Part 809.3(a)) to state that IVDs are devices under the Federal Food Drug & Cosmetic Act (FD&C Act), “including when the manufacturer of these products is a laboratory.”
Is This the End of the FDA’s Enforcement Discretion Policy on LDTs?
While the final rule ends the FDA’s enforcement discretion policy for most LDTs, it will continue to apply enforcement discretion to select LDTs in a general and targeted manner. For example, the FDA intends to continue the general enforcement discretion approach and generally not enforce any applicable requirements for the following tests:
“1976-Type LDTs”
Tests with the following characteristics that were common among LDTs offered in 1976:
-
- Use of manual techniques (without automation) performed by laboratory personnel with specialized expertise
- Use of components legally marketed for clinical use
- Design, manufacture, and use within a single CLIA-certified laboratory that meets the requirements under CLIA for high complexity testing
Certain Human Leukocyte Antigen (HLA) Tests for Transplantation
HLA tests that are designed, manufactured, and used within a single laboratory certified under CLIA that meets the requirements to perform high-complexity histocompatibility testing when used in connection with organ, stem cell, and tissue transplantation to perform HLA allele typing, for HLA antibody screening and monitoring or for conducting real and “virtual” HLA crossmatch tests.
Forensic Tests
Tests intended solely for forensic (law enforcement) purposes
Department of Defense (DoD) and Veterans Health Affair (VHA) LDTs
LDTs manufactured and performed within the DoD or VHA. This policy applies only to LDTs used for patients that are being tested and treated within the DoD or VHA.
The Following Tests Previously Excluded, Must Comply with the Following FDA Device Regulations:
-
- Tests intended as blood donor screening or human cells, tissues, and cellular and tissue-based products (HCT/P) donor screening tests required for infectious disease testing, or needed for the determination of blood group and Rh factors
- Tests intended for emergencies, potential emergencies, or material threats declared under section 564 of the FD&C Act
- Direct-to-consumer tests intended for consumer use without meaningful involvement by a licensed healthcare professional
Tests Manufactured and Offered exclusively for Public Health Surveillance are Unaffected by the Phaseout Policy.
-
- Furthermore, targeted enforcement discretion will also be applied by the FDA for certain LDTs. These targeted areas of enforcement discretion are best considered together with the agency’s phaseout policy.
LTD Phaseout Policy
Under the final rule, the FDA is phasing out general enforcement for LDTs in the following five stages beginning from the dates indicated below:
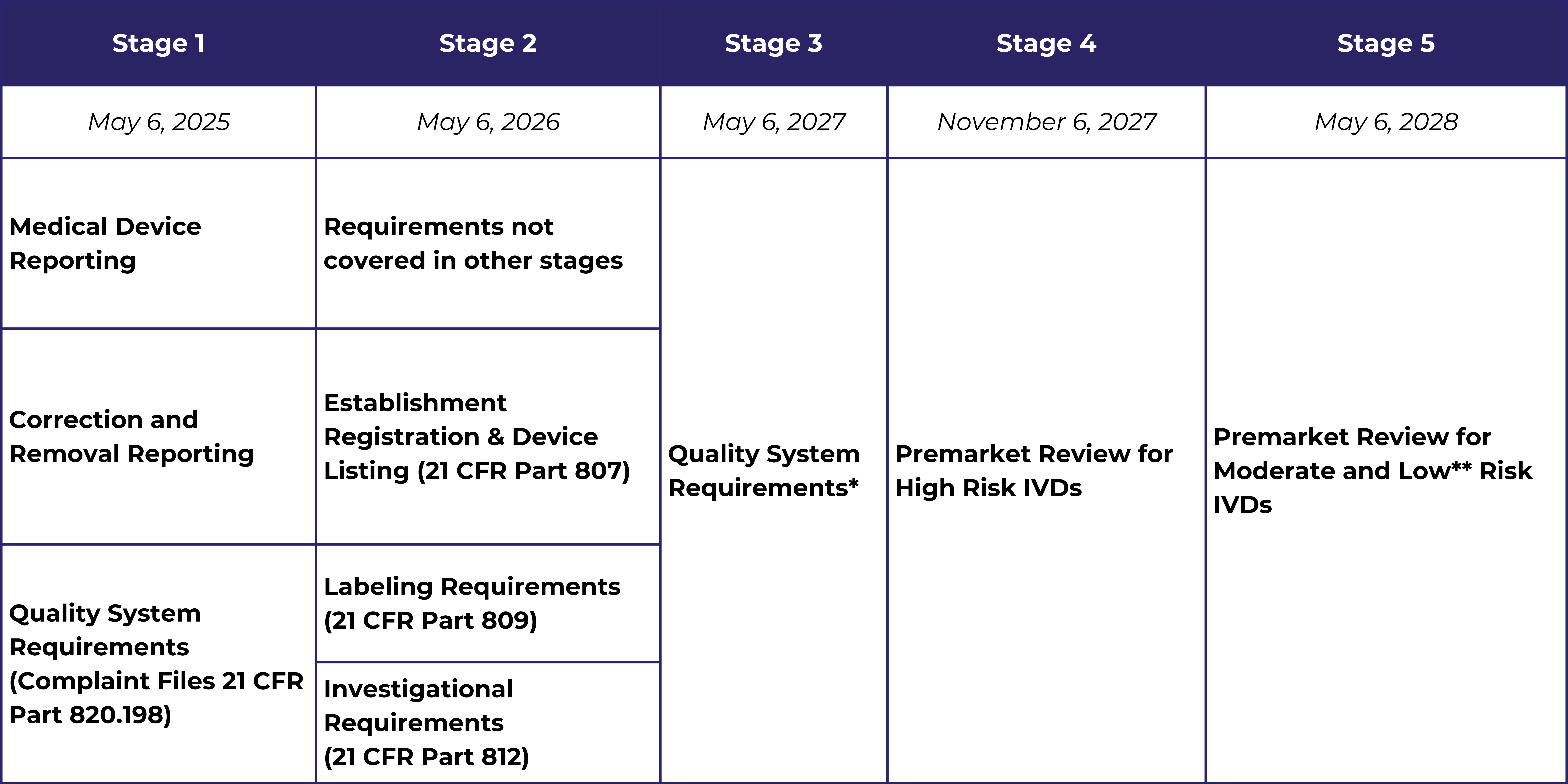
*For LDTs, FDA expects compliance with:
-
- Design controls (21 CFR Part 820.30)
- Purchasing controls (including supplier controls) under 21 CFR Part 820.50
- Acceptance activities (receiving, in-process, and finished device acceptance) under 21 CFR Parts 820.80 & 820.86
- CAPA under 21 CFR Part 820.100
- Records requirements under 21 CFR 820, subpart M
**Most low risk IVDs are exempt from premarket review
(21 CFR Part 807) (21 CFR Part 809) (21 CFR Part 812)
FDA LDT Timeline
Regarding the quality system requirements described above, these are references to the current quality system requirements established in the Quality System Regulation (QSR), 21 CFR Part 820. For laboratories, these requirements should also be considered within the context of the Quality Management System Regulation (QMSR) final rule published on January 31, 2024. This final rule becomes effective two years after its publication (2026) and will begin to be enforced by the FDA from February 2, 2026.
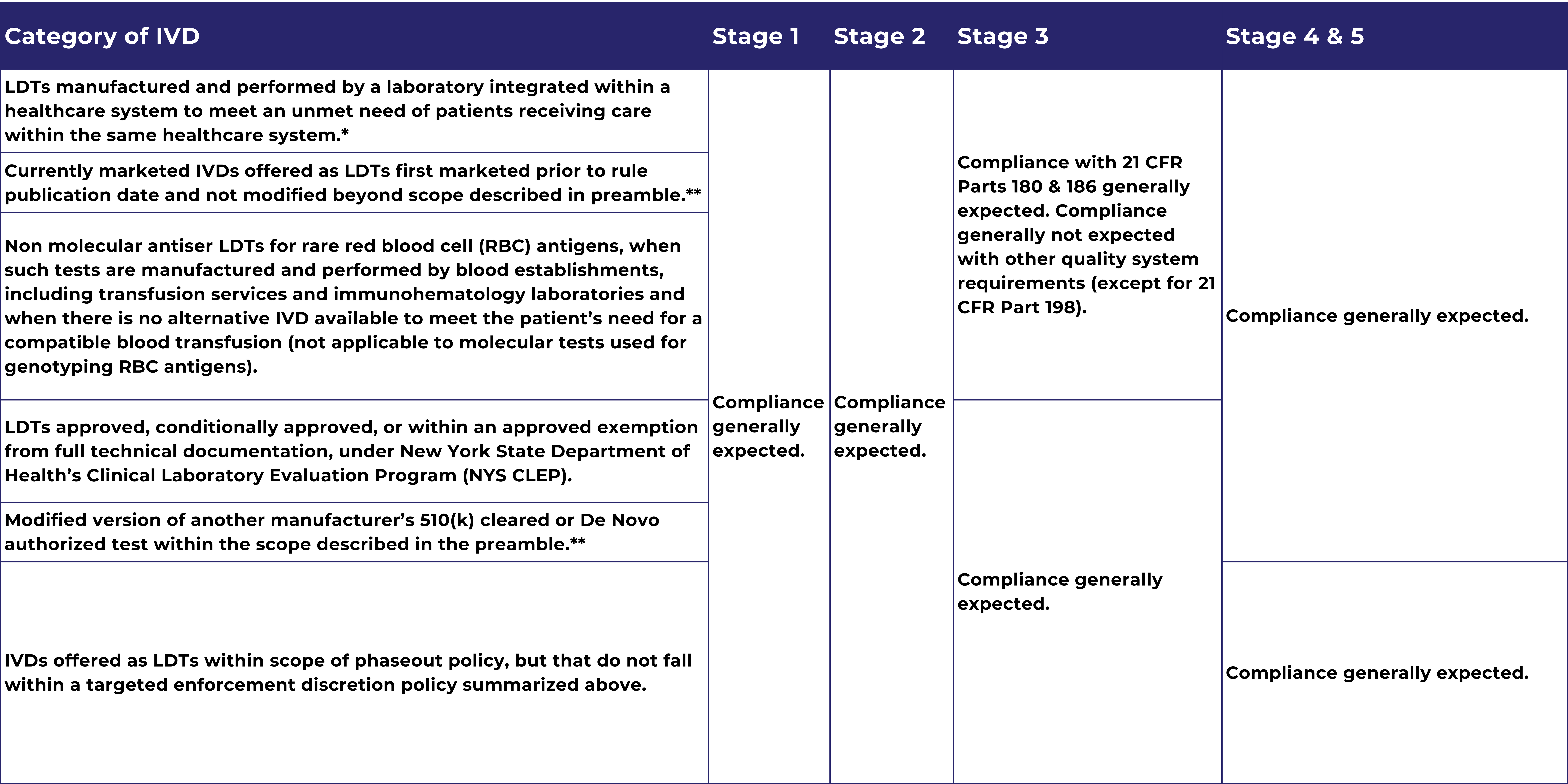
*FDA considers an LDT to be for an unmet need where there is no available FDA-authorized ID that meets the patient’s needs which may be because: (1) there is no FDA-authorized ID for the disease or condition; (2) there is an FDA-authorized IVD for the disease or conditions but it is not indicated for use on the patient or a unique attribute needs to be added to the LDT to meet the patient’s needs; (3) there is an FDA-authorized IVD but it is not available to the patient. The unmet needs LDT policy applies only to LDTs that are validated.
**The scope of changes described in the preamble for which premarket review and quality system requirements would be expected include: (1) Change to the indications for use of the IVD; (2) Alteration of the operating principle of the IVD (e.g. changes in critical reaction components); (3) Include significantly different technology in the ID (e.g. addition of artificial intelligence or machine learning to a test algorithm); (4) Adversely changing the performance or safety specifications of the IVD.
New York State Department of Health’s Clinical Laboratory Evaluation Program (NYS CLEP).
While further information and guidance regarding this final rule are expected from the FDA, it’s imperative that laboratories that manufacture LDTs marketed in the US already begin to assess the impact of this final rule on their products and plan for their transition.
Understanding FDA’s Final Ruling on Laboratory Developed Tests with MedEnvoy
This article provides an overview of the FDA’s final rule including a phaseout policy regarding LDTs. Should you have any questions regarding these requirements or require further US regulatory consulting support or a US Agent, get in touch.