Home » Resources & News » FDA Human Factor in Medical Device Risk Management
Using any type of product in a manner not intended by its manufacturer has the potential to cause harm, a risk that is closely examined under FDA Human Factor considerations. In the case of medical devices, due to their therapeutic or diagnostic purposes, the nature of such harm may be significant. This type of use is referred to as “off-label” use. Since medical devices may be used by the lay public or appropriately qualified healthcare professionals, the type of user also plays a significant role in the severity and/or likelihood of harm occurring.
To learn more about how you can streamline your medical device to the US market, click here.
The Importance of Addressing FDA Human Factors in Medical Device Risk Management
For these reasons, among others, the consideration of reasonably foreseeable misuse of medical devices is critical when performing risk management. In fact, it is a requirement under ISO 14971 that manufacturers document reasonably foreseeable misuse, which the standard defines as: “Use of a product or system in a way not intended by the manufacturer but which can result from readily predicted human behavior.”
Within the U.S. regulatory framework, the term “use error” is defined similarly to other global standards. According to current FDA guidance, a use error is:
“User action or lack of action that was different from that expected by the manufacturer and caused a result that (1) differed from the result expected by the user, (2) was not caused solely by device failure, and (3) did or could result in harm.”
While the level of risk associated with a particular hazard (e.g., a hazard related to a specific use error) is typically determined based on the severity and probability of potential harm, assessing the likelihood of use errors can be challenging. Some use errors may not be reasonably foreseeable until intended users have had an opportunity to use the device (e.g., in simulated use) and be observed. Therefore, the severity of potential harm is often more meaningful in determining the risk controls necessary to eliminate or reduce that harm.
For this reason, the FDA considers human factors engineering (HFE) and usability testing critical components of medical device design and development. The FDA’s Center for Devices and Radiological Health (CDRH) maintains that, for devices where risk analysis indicates the possibility of serious harm to patients or users due to use errors, manufacturers should apply appropriate human factors or usability engineering processes. This applies especially when devices are modified to address design deficiencies related to device use.
What are Human Factors Engineering and Usability Testing?
Current FDA guidance defines human factors engineering as:
“The application of knowledge about human behavior, abilities, limitations, and other characteristics of medical device users to the design of medical devices, including mechanical and software-driven user interfaces, system tasks, user documentation, and user training, to enhance and demonstrate safe and effective use. Human factors engineering and usability engineering can be considered synonymous.”
Users may interact with devices in various ways, depending on the nature of the device’s output (e.g., visual, aural, or tactile information) and how data or information is input into the device. All points of interaction between the user and the device, including all elements with which the user interacts, are part of the “user interface.” The “user interface” also includes all sources of information transmitted by the device, such as packaging, labeling, training, and all physical controls and display elements (including alarms and the logic of operation of each device component and the user interface system).
The focus of HFE/usability testing is on studying and understanding how individuals interact with technology and how user interface design affects those interactions, as well as considering human factors and the corresponding outcomes shown in the figure below:
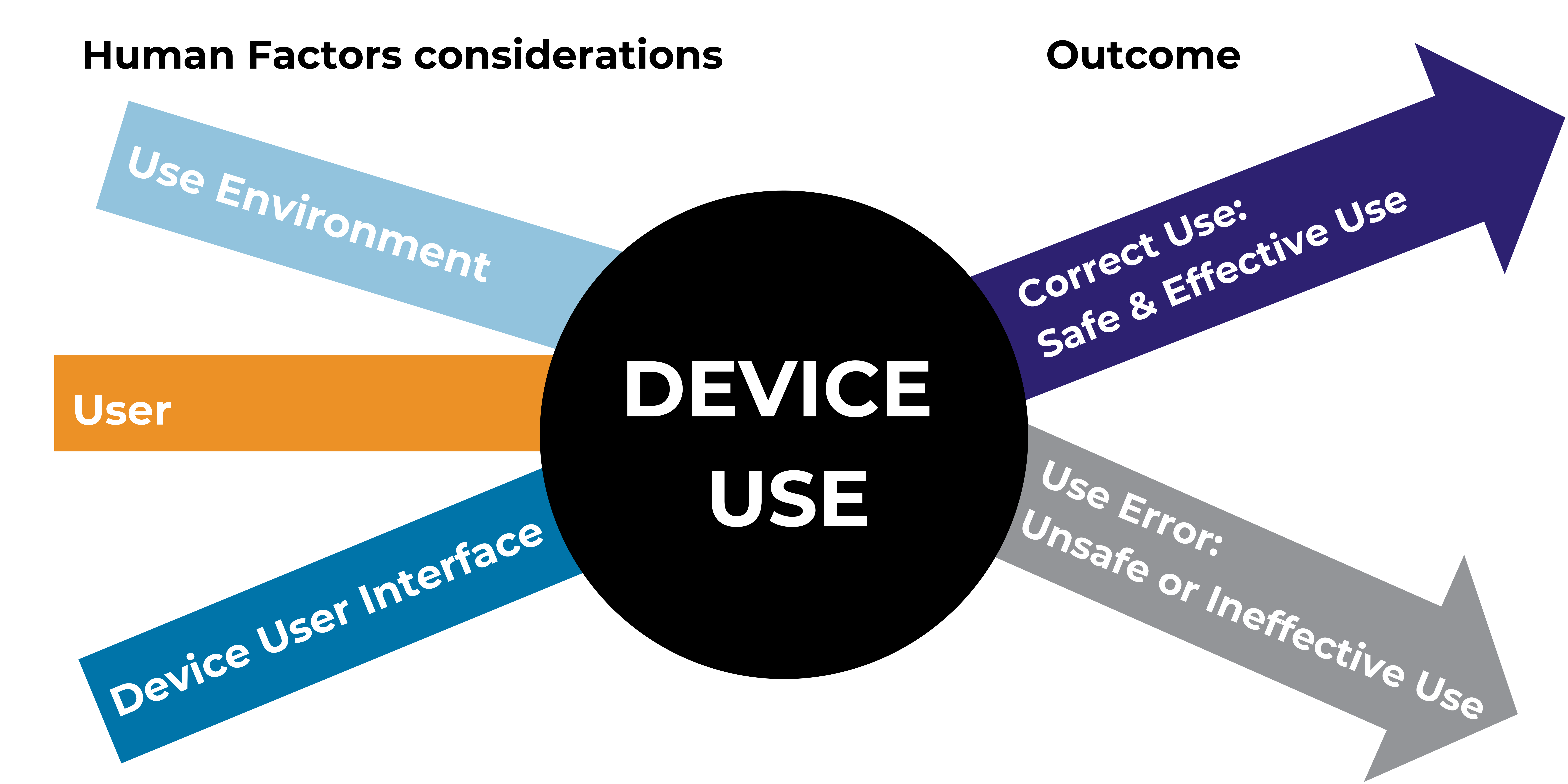
Figure 1 – Device Use and Human Factor Considerations: Inputs and Corresponding Outputs
(Source: FDA Guidance – Applying Human Factors and Usability Engineering to Medical Devices (February 3, 2016))
Testing performed at the end of device design and development to assess user interactions with a device’s user interface and identify user errors that could result in serious harm to the patient or user is defined in FDA guidance as “human factors validation testing.” This testing is also used to assess the effectiveness of risk controls and represents one portion of design validation. Additionally, the outputs of this testing may inform required warnings or precautions to be added to labeling to prevent potential misuse.
Device Users, Use Environments, and User Interface
It is critical that medical devices be used by their intended users without errors that could cause harm to themselves or the patients using the devices. The FDA defines a user as:
“A person who interacts with (i.e., operates or handles) the device.”
As mentioned previously, devices may have different types of users, such as healthcare professionals or laypersons. Before performing HFE analyses, and as part of the risk management process, manufacturers should ensure the following user information is established:
Type of intended user: (e.g., healthcare professionals, nursing staff, caregivers, patients, etc.).
User characteristics: Factors that could impact device safety and effectiveness (e.g., physical, sensory, or cognitive abilities; education, knowledge, and experience; behaviors; age, etc.).
Training Requirements: The necessary level of training users are expected to have or receive before using the device.
Considerations for the device’s user interface should cover all stages of device use, including setup (e.g., unpacking, installation, calibration), intended use, and maintenance or technical support (e.g., cleaning, reprocessing, replacing, or repairing parts/components). Device interface characteristics to consider may include, among others:
-
- Software-related characteristics such as graphical user interfaces.
- Alarms, indicators, and displays or other elements that provide information to the user.
- Physical characteristics (e.g., device size and shape).
- Interoperability with other devices or products.
- Hardware components with which the user interacts (e.g., switches, buttons, knobs).
Medical devices, particularly those intended for lay use, may be used in different environments subject to a range of conditions (e.g., lighting, temperature, humidity, noise, distractions). Such environments may include special settings such as intensive care units (where many devices are used simultaneously), homes, or even outdoor environments. As part of HFE and risk management, manufacturers should establish the intended use environments and design their devices so that their safety and effectiveness are not adversely affected by such conditions.
Preliminary FDA Human Factors Engineering Analyses and Evaluations
There is significant overlap between the tools commonly used in preliminary HFE analyses and evaluations and those used in risk management, such as Failure Mode Effects Analysis (FMEA) and Fault Tree Analysis (FTA) to identify and categorize critical tasks. Additionally, publicly available information, such as the FDA’s Total Product Life Cycle (TPLC) database, can be used to identify reasonably foreseeable misuse or use errors.
However, HFE also employs analytical (reviewing and assessing user-device interactions) and empirical (involving user experiences with the device, prototypes, or mock-ups) approaches to identify critical tasks, including:
Analytical approaches:
-
- Task analysis
- Heuristic analysis
- Expert review
Empirical approaches:
-
- Contextual inquiry
- Interviews
- Formative evaluations (used to inform the device’s user interface design during development)
- Cognitive walkthrough
- Simulated-use testing
Formative evaluations complement and refine the analytical approaches used, leading to further refinements of the device’s risk management file. Formative evaluations should be controlled within the framework of the manufacturer’s quality management system (QMS) and performed according to a pre-established protocol, which includes:
-
- Evaluation purpose, goals, and priorities.
- Description of the user interfaces to be assessed.
- Use scenarios and tasks involved.
- Evaluation participants.
- Data collection methods.
- Data analysis methods.
- Description of how the evaluation results will inform design and development.
The FDA HFE guidance provides overviews of these approaches and their applications in HFE, including sample templates for HFE Reports, considerations for determining sample sizes for human factors validation testing, analyzing testing results, and relevant references.
FDA Human Factors Engineering and Actual Use Testing
In some cases, usability testing may be necessary under actual rather than simulated use conditions (e.g., prosthetics). Whenever actual use testing is required, it should always be preceded by simulated use testing to ensure that device design sufficiently addresses safety under simulated conditions. If actual conditions are necessary to determine safety and effectiveness, and the requirements under 21 CFR Part 812 apply, an Investigational Device Exemption (IDE) will be required. Such testing requires that the test participants and the clinical environment be representative of the intended users and use environments.
While actual use testing may be conducted in the context of a clinical study, these studies are more closely supervised than real-world testing, and participants are generally trained differently. Therefore, care should be taken to ensure that the results of actual use testing are representative of real-world device use.
This article provides an overview of current US FDA human factors and usability guidance. If you have any questions regarding US FDA regulatory support, representation, or risk management consulting, feel free to get in touch. To contact our team, click here. To learn more specifically about our FDA service, click here.
Related Resources



Related Guidance
We share our expertise because we believe in removing barriers to global commercialization.
5 mins
How Do US Medical Device Executives Build a Business Case for Going Global?
Learn how medical device executives calculate ROI, navigate regulatory costs, and build compelling global expansion business cases.
6 mins
How Does Global Population Aging Create Opportunities for US Medical Device Exporters?
Aging populations in Japan, Germany, and China drive massive demand for US medical devices across cardiovascular, diagnostic, and diabetes categories.
6 mins
What Does a Successful International Launch Look Like for a US Medical Device Company?
Learn proven strategies for successful international medical device launches, including regulatory planning and market selection insights.