
ISO 13485 Medical Devices: Global Regulatory Compliance
ISO 13485:2016 standards play a fundamental role in global regulatory compliance for medical devices. Nearly 30 years after the initial version of this standard was published in 2003, its influence has expanded into markets that traditionally viewed it as misaligned with their own Good Manufacturing Practice (GMP) regulations.
In this article, we provide an overview of the role of this standard in both EU and US regulatory frameworks, as well as its significance in the Medical Device Single Audit Program (MDSAP).
For information on MedEnvoy’s EU services, click here. For information on MedEnvoy’s US services, click here.
ISO 13485 Within the EU Regulatory Framework
With the introduction of the revised medical device regulatory frameworks under the MDR/IVDR, particularly in the case of the IVDR with increased Notified Body involvement in manufacturer conformity assessment, more robust quality management system (QMS) requirements were established. Article 10 and Annexes IX and XI of the MDR/IVDR set out several general and specific QMS requirements, including the establishment of procedures/processes for clinical/performance evaluation, risk management, post-market surveillance (PMS), and unique device identification (UDI). While ISO 13485 for medical devices requires organizations to document and maintain the effectiveness of their QMS in accordance with applicable regulatory requirements, it does not provide detailed provisions regarding such procedures/processes.
Harmonized Standards and Presumption of Conformity
Conformity with standards harmonized under the MDR/IVDR is not mandatory; however, there is a presumption of conformity with relevant provisions of the regulations when these standards are applied. While EN ISO 13485:2016 for medical devices is harmonized under the MDR/IVDR, the ZA, ZB, and ZC annexes that demonstrate the relationship between the standard and the AIMDD/MDD/IVDD do not adequately allow organizations to determine the relationship between the standard and the MDR/IVDR.
Amendments to EN ISO 13485:2016
In 2021, EN ISO 13485:2016 for medical devices was amended to EN ISO 13485:2016 + A11:2021, which is also harmonized under the MDR/IVDR. In this amended version, the previous Z annexes that demonstrated the relationship between the standard and device-related directives were replaced with new ZA and ZB annexes, demonstrating the relationship between the standard and the MDR/IVDR. These annexes provide information on whether the MDR/IVDR requirements are fully, partially, or not covered by the standard across three separate tables, which cover the general manufacturer obligations under Article 10 and the relevant conformity assessment-related requirements in Annexes IX and XI of the regulations.
Importance of Gap Assessments
An important point in the Z annexes of the amended standard is that where definitions established in EN ISO 13485 differ from specific definitions established in the MDR/IVDR, for the purposes of EU regulatory compliance, the definitions in the regulations take precedence. This illustrates the importance for manufacturers with ISO 13485-compliant QMSs to perform gap assessments of their systems against these Z annexes to ensure all potential gaps are addressed when transitioning to the MDR/IVDR. Considering the extended transition timeframes under the MDR/IVDR, which require manufacturers to implement MDR/IVDR-compliant QMSs, it is critical that transitioning organizations prioritize such assessments.
ISO 13485 Within the U.S. Regulatory Framework
Manufacturers marketing their medical devices in the U.S. and its territories should already be familiar with FDA GMP requirements established under 21 CFR Part 820, also known as the Quality System Regulation (QSR), even if their devices are exempt from such requirements based on their classification.
With its own GMP requirements having been in place since 1978 (and subject to a major revision that went into effect in 1997), the FDA has historically been unwilling to adopt other GMP/QMS requirements for devices and had a particular disdain for the CE marking regulatory framework, largely due to the commercial relationship between manufacturers and Notified Bodies. As such, until recently, the FDA was reluctant to recognize the ISO 13485 medical devices standard. However, while ISO 13485 is still not a voluntary consensus standard recognized by the FDA, the agency’s stance on this standard has evolved over time, particularly due to its involvement in MDSAP and efforts toward regulatory harmonization, which aligns with the agency’s “least burdensome” approach.
The Quality Management System Regulation (QMSR) Final Rule
On January 31, 2024, the FDA issued the Quality Management System Regulation (QMSR) Final Rule, which amends 21 CFR Part 820 by incorporating ISO 13485:2016 by reference. This rule becomes effective on February 2, 2026, after which the FDA will enforce QMSR requirements. While manufacturers must continue to comply with the current QSR until February 2, 2026, they should begin preparing for the transition to the QMSR.
Recommendations for Manufacturers
First, it is recommended that manufacturers (if they have not already done so) familiarize themselves with the changes to 21 CFR Part 820 detailed in the final rule. For combination product manufacturers, this also includes understanding the changes to 21 CFR Part 4, which are also detailed in the final rule. The changes to 21 CFR Part 820 can be found here.
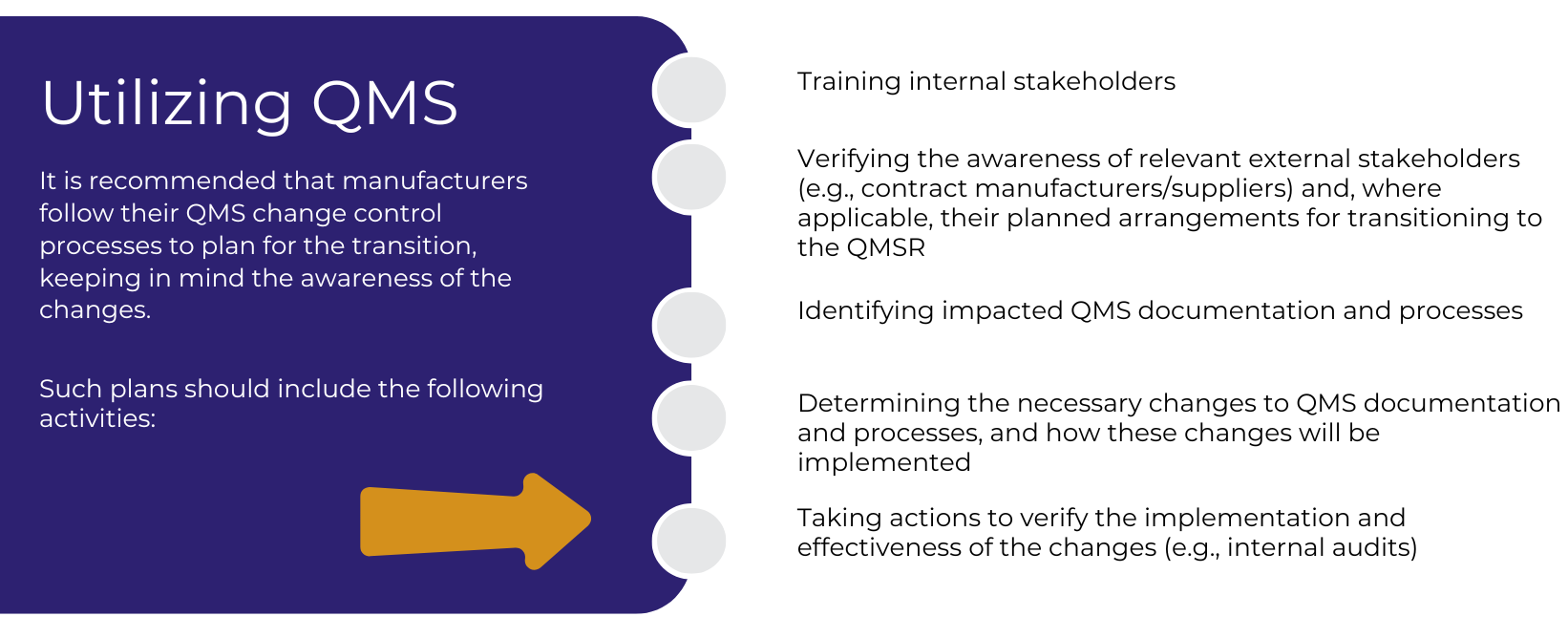
Once the transition is complete, manufacturers must ensure ongoing compliance with the QMSR, as the FDA is already investing in the preparation and training of its inspectors for the transition. By February 2, 2026, all relevant establishments will be inspected for compliance with the QMSR.
ISO 13485 and its Role Within MDSAP
It has been over ten years since the MDSAP pilot was rolled out, and during that time, the MDSAP program has undergone several improvements, with increased interest from various regulatory agencies in leveraging the program for their own regulatory frameworks. Despite these changes, ISO 13485 continues to serve as the backbone of the MDSAP audit approach, as established in MDSAP AU P0002.009, complemented by the respective regulatory requirements of Australia, Brazil, Canada, Japan, and the United States.
Compliance with ISO 13485 and the applicable regulatory requirements is essential for manufacturers seeking MDSAP certification, as they will be audited to these requirements. The relevant MDSAP chapters and their respective tasks across the ISO 13485 clauses can be found here.
The only tasks from the MDSAP audit approach not listed in the above matrix are those that require the selection of specific files for review:
-
- Chapter 2 – Design and Development, Task 2 – Selection of a completed design and development project
- Chapter 6 – Production and Service Controls, Task 2 – Selection of production and service process(es)
- Chapter 7 – Purchasing, Task 2 – Selection of a supplier file to audit
Stay up to Date on the Role of ISO 13485 in Global Regulatory Compliance for Medical Devices with MedEnvoy
This article provides an overview of the role of ISO 13485 in global regulatory compliance for medical devices. Whether you have questions about the QMS implementation, auditing or training services, or have questions on global regulatory compliance, we are here to help. To connect with one of our regulatory experts and discuss your unique needs, click here.


