
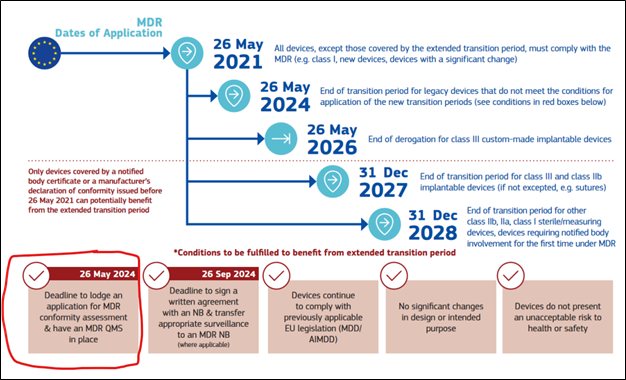
The Medical Device Regulations (2017/745) (“MDR”) called out a Date of Application of 26 May 2021, for which all Class I devices, which are not sterile or measuring, or subject to reclassification, e.g., reusable surgical devices, were required to fully comply with the requirements of the MDR. This included the requirements of Article 10(9), as it relates to a quality management system. With the introduction of Regulation (EU) 2023/607, the European Commission’s timeline for transition to the MDR for higher risk class (“Legacy”) medical devices was revised to 31 Dec 2027 (all Class III devices and select Class IIb devices) and 31 Dec 2028 (remaining Class IIb devices, Class IIa devices and Class I – sterile or measuring). However, the 2023/607 regulation specifically called out the requirement that all Legacy devices would be subject to the quality management system requirements detailed in Article 10(9) of the MDR as of 26 May 2024.
Preparing for Compliance: Deadline Approaching
Manufacturers of Legacy devices that have delayed the transition of their devices to the MDR must have updated any existing quality management system to comply with Article 10(9), and if they had not implemented a quality management system previously, this must be completed by 26 May 2024; otherwise, these manufacturers will not be permitted to extend the transition to the MDR as outline in regulation 2023/607.
What are these quality management system requirements from Article 10(9)? As shown below, these requirements look to expand upon the requirements of ISO 13485:2016, to include the specific elements of the MDR. As an example, ISO 13485:2016 §8.5.1 references the necessity to conduct postmarket surveillance; however, the ISO 13485 standard doesn’t indicate what the manufacturer should include in that process. MDR Article 10(9)(i), which references Article 83 and Annex III, provides a thorough description of the requirements for a post-market system. Additionally, while ISO 13485:2016 §4.1.1 identified that manufacturers shall fulfill the requirements of “applicable regulatory requirements”, MDR Article 10(9)(a) identifies the necessity to have procedures to address the conformity assessment process for medical devices, e.g., ensuring that there is a documented processes for developing Technical Documentation (file) per Annex II of the MDR.
If you are a manufacturer of a Legacy medical device the 26 May 2024 deadline is quickly approaching. Have you implemented the necessary modifications to your quality management system? If you are questioning your current status, contact MedEnvoy Global BV who can assist with performing a gap analysis, procedure updates or a compliance review. If you are a manufacturer of a Class I manufacturer and you have not considered the quality management system requirements of MDR Article 10(9), let MedEnvoy help you.
MDR Article 10(9) calls out the need to include the following in your quality management system. For your reference we have included the applicable section of ISO 13485:2016 that links most closely to the subsections of Article 10(9).
| Article 10 General Obligations of Manufacturers, Subsection 8 | Reference in ISO 13485 |
| (a) a strategy for regulatory compliance, including compliance with conformity assessment procedures and procedures for management of modifications to the devices covered by the system; | ISO 13485 §4.1 General Requirements and ISO 13485 §7.3.9 Control of Design and Development Changes |
| (b) identification of applicable general safety and performance requirements and exploration of options to address those requirements; | ISO 13485 §7.3 Design and Development. Consider MDR Annex I |
| (c) responsibility of the management; | ISO 13485 §5 Management Responsibility |
| (d) resource management, including selection and control of suppliers and sub-contractors; | ISO 13485 §7.4 Purchasing |
| (e) risk management as set out in Section 3 of Annex I; | ISO 13485 §7.1 Planning of Product Realization, ISO 14971 |
| (f) performance evaluation, in accordance with Article 61 and Annex XIV, including PMCF; | ISO 13485 §7.3.7 Design and Development Validation |
| (g) product realisation, including planning, design, development, production and service provision; | ISO 13485 §7.3 Design & Development. |
| (h) verification of the UDI assignments made in accordance with Article 27(3) to all relevant devices and ensuring consistency and validity of information provided in accordance with Article 29; | ISO 13485 §7.5.8 Identification |
| (i) setting-up, implementation and maintenance of a post-market surveillance system, in accordance with Article 83; | ISO 13485 §8.5 Improvement. Consider MDR Annex III. |
| (j) handling communication with competent authorities, notified bodies, other economic operators, customers and/or other stakeholders; | ISO 13485 §7.2.3 Communications |
| (k) processes for reporting of serious incidents and field safety corrective actions in the context of vigilance; | ISO 13485 §8.2.3 Reporting to Regulatory Authorities and §8.3.3 Actions in Response to Nonconforming Product Detected after Delivery |
| (l) management of corrective and preventive actions and verification of their effectiveness; | ISO 13485 §8.5.2 Corrective Action and §8.5.3 Preventive Action |
| (m) processes for monitoring and measurement of output, data analysis and product improvement. | ISO 13485 §8.4 Data Analysis |
https://health.ec.europa.eu/system/files/2023-08/timeline_mdr_en.pdf
QMS Requirements Under IVDR
Within the current In Vitro Diagnostic Device Regulations (2017/746) (“IVDR”), Class A devices must fully comply with the requirements of Article 10(8), as it relates to a quality management system, by the Date of Application of the IVDR, which was 26 May 2022. For all other IVDs, including Class A (sterile), Class B, Class C and Class D, the deadline for implementation of an IVDR-compliant quality management system is 26 May 2024.
On 23 Jan 2024 the European Commission issued a proposal[1] to amend Regulations (EU) 2017/745 and (EU) 2017/746, which proposes changes to the transitional provisions for certain in vitro diagnostic medical devices. Within the document the European Commission proposes to amend Article 110 (Transitional Provisions) such that the deadline for higher risk class IVDs to comply with all aspects of the IVDR is delayed to 31 Dec 2027 (class D devices), 31 Dec 2028 (class C devices), and 31 December 2029 (class B devices and for class A – sterile). One of the conditions for IVD manufactures that wish to extend the timeline for IVDR compliance is that these manufacturers must have a quality management system in place that addresses the requirements of Article 10(8) by 25 May 2025. For these manufacturers they will be required to follow the Conformity Assessment routes detailed in Annex IX or Annex XI, for which they will most often have an ISO 13485 quality management system, which is certified by their Notified Body. With the recent proposal of the European Commission, the manufacturers will need to have their Notified Body evaluate their quality management system for the requirements of ISO 13485, as well as the requirements of Article 10(8).
As it relates to Article 10(8), the IVDR calls out that manufacturers of devices[2] must establish, document, implement, maintain, keep up to date and continually improve a quality management system, which covers all parts and elements of a manufacturer’s organization dealing with the quality of processes, procedures and devices. Specifically, Article 10(8) calls out the need to address the following:
| Article 10 General Obligations of Manufacturers, Subsection 8 | Reference in ISO 13485 |
| (a) a strategy for regulatory compliance, including compliance with conformity assessment procedures and procedures for management of modifications to the devices covered by the system; | ISO 13485 §4.1 General Requirements and ISO 13485 §7.3.9 Control of Design and Development Changes |
| (b) identification of applicable general safety and performance requirements and exploration of options to address those requirements; | ISO 13485 §7.3 Design and Development. Consider IVDR Annex I |
| (c) responsibility of the management; | ISO 13485 §5 Management Responsibility |
| (d) resource management, including selection and control of suppliers and sub-contractors; | ISO 13485 §7.4 Purchasing |
| (e) risk management as set out in Section 3 of Annex I; | ISO 13485 §7.1 Planning of Product Realization, ISO 14971 |
| (f) performance evaluation, in accordance with Article 56 and Annex XIII, including PMPF; | ISO 13485 §7.3.7 Design and Development Validation |
| (g) product realisation, including planning, design, development, production and service provision; | ISO 13485 §7.3 Design & Development. |
| (h) verification of the UDI assignments made in accordance with Article 24(3) to all relevant devices and ensuring consistency and validity of information provided in accordance with Article 26; | ISO 13485 §7.5.8 Identification |
| (i) setting-up, implementation and maintenance of a post-market surveillance system, in accordance with Article 78; | ISO 13485 §8.5 Improvement. Consider IVDR Annex III. |
| (j) handling communication with competent authorities, notified bodies, other economic operators, customers and/or other stakeholders; | ISO 13485 §7.2.3 Communications |
| (k) processes for reporting of serious incidents and field safety corrective actions in the context of vigilance; | ISO 13485 §8.2.3 Reporting to Regulatory Authorities and §8.3.3 Actions in Response to Nonconforming Product Detected after Delivery |
| (l) management of corrective and preventive actions and verification of their effectiveness; | ISO 13485 §8.5.2 Corrective Action and §8.5.3 Preventive Action |
| (m) processes for monitoring and measurement of output, data analysis and product improvement. | ISO 13485 §8.4 Data Analysis |
[1] Proposal for a REGULATION OF THE EUROPEAN PARLIAMENT AND OF THE COUNCIL amending Regulations (EU) 2017/745 and (EU) 2017/746 as regards a gradual roll-out of Eudamed, information obligation in case of interruption of supply and the transitional provisions for certain in vitro diagnostic medical devices… [Brussels, 23.1.2024; COM(2024) 43 final; 2024/0021 (COD)] – https://health.ec.europa.eu/document/download/bcde7f36-b2fe-4d5f-989a-6daa80538b79_en?filename=mdr_in-vitro-proposal.pdf
[2] The requirements of Article 10(8) do not apply to manufacturers of devices for performance study.
If you have any questions or need assistance meeting the QMS deadline, please contact us.