Saudi Arabia Medical Device Registration Requirements
The Saudi Food and Drug Administration (SFDA) has aligned its medical device regulatory requirements with other major market frameworks, which makes it an attractive expansion market for companies who already have European CE Marking, for instance. However, it is helpful to understand the process up front since there are some differences. In this article, we will cover steps in the Saudi Arabia medical device registration process for foreign manufacturers.
Saudi Arabia Regulatory Framework for Medical Devices
The Saudi Arabia medical device registration process is regulated by the SFDA according to the Medical Devices and Supplies Regulation (link in Arabic), which went into effect in 2021. The regulation conveys the most requirements from the Medical Device Interim Regulations, which established that medical devices must obtain Medical Device Marketing Authorization (MDMA) from the SFDA before being distributed in Saudi Arabia. There are exceptions for devices that are considered safe and effective and for devices not intended for commercial use. If your device is novel or innovative, SFDA may authorize exemptions from certain steps in the registration process.
All devices that require MDMA must now follow the Technical File Assessment (TFA) route. Technical files for authorizations from Global Harmonization Task Force (GHTF) member countries (EU, USA, Canada, Japan and Australia) are no longer accepted as reference. However, prior authorization in one GHTF market is a prerequisite for MDMA certification.
Your technical file and registration documentation must be submitted through the Unified Electronic System (GHAD), which is SFDA’s online portal. If your company is not based in Saudi Arabia, you must appoint an in-country representative known as an Authorized Representative (AR) to submit your MDMA application on your behalf.
Choosing an In-Country Representative in Saudi Arabia
As a foreign manufacturer, your AR plays a critical role in the registration and distribution of your device. They will be your liaison with SFDA and carry out essential activities, including:
-
- Submit documentation to the SDFA for Medical Device Market Authorization (MDMA) and company registration.
-
- Your AR’s name, address, and contact information will be printed on your device’s technical documentation, labeling, packaging, and IFU.
-
- Maintain post market compliance and vigilance, such as reporting device modifications and adverse events to SFDA.
-
- Maintain your device registration by submitting renewal applications as needed.
Your AR can be an individual or company located in Saudi Arabia with a Medical Device Establishment License (MDEL). Many manufacturers consolidate the AR role by designating a distributor as their representative. However, there are long-term benefits to contracting an independent company that only offers authorized representative services. For example, your AR effectively owns and controls the MDMA certification for your device, so changing or adding distributors can be complicated if your distributor is your AR. An independent party gives you full control over your registration and the freedom to make any marketing or distribution decisions without delay or conflicts of interest.
Saudi Arabia Medical Device Registration and Classification
Medical devices are classified according to a set of rules that are informed by the European Medical Devices Regulation (EU MDR 2017/745) and GHTF. Rules for medical devices and IVDs are published in Annex 5 of MDS-REQ1: Requirements for Medical Device Marketing Authorization. MDS – G008: Guidance on Medical Devices Classification provides in depth explanations of each rule.
Medical devices are classified into four levels of increasing risk, though low-risk devices are distinguished based on specific functions and characteristics:

Some devices that do not have an intended medical purpose, but that have specific cosmetic uses, are regulated as medical devices. Examples of these products include colored contact lenses, body contouring implants, and dermal fillers.
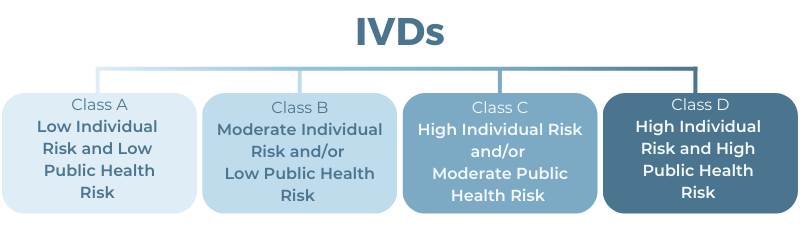
IVDs are Classified into Four Tiers of Increasing Risk: 
Grouping Criteria for Medical Devices and IVDs
Medical devices that share a common intended purpose, such as key indications, medical
conditions, and patient populations, can be bundled in the same MDMA application as long as they meet the following criteria:
-
- Same legal manufacturer,
- Same intended use/purpose,
- Same risk class,
- Same Global Medical Device Nomenclature (GMDN) code (optional), and
- Common physical design, construction material, and manufacturing process.
Medical Device Quality Management System (QMS) Requirements
Medical device manufacturers, importers, and distributors are required to implement and maintain a quality management system according to the Saudi QMS standard, SFDA.MD/GSO ISO 13485, or an equivalent. For manufacturers of Class A and B devices, a QMS certified from a Saudi Conformity Assessment Body (CAB) is required. For Class C and D devices, SFDA may also require an inspection or audit of the manufacturer’s QMS.
Technical Documentation File Requirements
A technical file for SFDA approval comprises many of the same requirements and sections as a European Technical Documentation File. Key sections include:
-
- Device description: In addition to identifying information, the device description should include drawings and diagrams, materials, intended use, indications for use, patient populations, intended users, and classification justification.
-
- Labeling and Instructions for Use (IFU): Information provided by the manufacturer includes labeling, Unique Device Identifier (UDI), packaging, instructions for use, as well as any promotional materials. This section should include a full set of labels and packaging, the IFU, and instructions for handling, storage, transportation, installation, and maintenance of the device.
-
- Design and Manufacturing information: An in-depth description of the device design, such as detailed technical drawings, technical specifications, materials, requirements documentation, design traceability, design history, and confirmation of the verification and validation conducted on the device. This section should also include detailed information about the manufacturing process, the name and address of the manufacturing site, and the identification of suppliers and subcontractors.
-
- Essential Principles of Safety and Performance: This section provides rationale that your device meets the applicable Essential Principles of Safety and Performance in Annex I. You should identify the principles that apply to your device and explain why other principles may not; describe the methods used to demonstrate your device’s conformity with applicable principles; any standards applied to demonstrate conformity; and identify controlled documents that serve as evidence.
-
- Benefit-Risk Analysis and Risk Management: Your risk assessment should demonstrate that the device is safe. This section should include your Risk Procedure, Risk Management Plan, Risk Analysis, and Risk Management Report.
-
- Product Verification and Validation: This section explains the methods, tests, and standards used to verify and validate your device, including detailed explanations of tests and/or clinical study design, Clinical Evaluation Report (CER), and Post Market Clinical Follow-up (PMCF) Plan and Report. If no testing was conducted, you must include justification for foregoing testing and/or PMCF.
-
- Post Market Surveillance (PMS) Plan: Your PMS plan should demonstrate that you have adequate systems in place to effectively collect, analyze, and respond to post market data. Systems should include device traceability and communication to economic operators and users regarding device safety and performance issues.
-
- Periodic Safety Update Report (PSUR) and Post Market Surveillance Report: Class A devices must maintain a Post Market Surveillance Report that summarizes post market data, findings and conclusions from PMS data analysis, and any corrective and preventive actions (CAPAs) taken. PMS reports should be updated as needed and available on request by the SFDA. Class B, C, and D devices must maintain a Periodic Safety Update Report, in lieu of a PMS report, that includes the same information as a PMS report as well as risk-benefit determination conclusions, PMCF results, and sales volume. Class C and D manufacturers must update their PSUR annually; Class B manufacturers should update it as needed but at least every two years.
The fundamental requirements for an IVD technical file are nearly identical to that of a medical device technical file. Key differences in documentation requirements can primarily be found in the Product Verification and Validation section, which addresses product characteristics that are specific to IVDs, as well as the Performance Evaluation and Post Market Performance Follow-up requirements.
Saudi Arabia MDMA Review Timelines and Approval Process
With your technical file complete, your AR will submit your MDMA application to SFDA via GHAD. SFDA will conduct an initial completeness review. If successful, you will pay the assessment fees, and your application will move on to the formal regulatory review. During the review process, SFDA may issue additional information requests and it’s important to respond quickly to avoid delays. The official MDMA review timeline is 35 working days. However, actual review times can take three to six months, or even longer for very high-risk devices and in cases where additional information is requested.
On approval, SFDA will issue an MDMA certification in English and Arabic. The certificate includes the Medical Device National Listing Number, certificate number, and validity period, which is three years for most devices.
Take the Next Step Toward SFDA Medical Device Registration
The Saudi Arabia medical device registration process and market is straight forward for manufacturers who already have approval in a GHTF market. Foreign manufacturers still need an experienced In-Country Saudi Arabia Authorized Representative and, while there are many similarities between an SFDA and European technical file, many manufacturers need expert help adapting their documentation to SFDA requirements. Through our partners in Riyadh, MedEnvoy can act as your Saudi Arabia Authorized Representative. Our regulatory experts can ensure your technical file and registration documentation are ready for SFDA review. Contact us with your questions about Saudi Arabia.


