
The United Kingdom Medicines and Healthcare products Regulatory Agency (MHRA), under the jurisdiction of the UK Department of Health and Social Care, is responsible for regulating medical devices in the UK. For manufacturers, understanding MHRA requirements is essential to ensuring compliance with Great Britain Medical Device Registration standards.
Navigating Medical Device Registration in Great Britain
It is crucial for device manufacturers to understand that different regulatory frameworks apply within the UK, depending on whether a device is marketed or put into service in Great Britain or Northern Ireland, each with its own specific regulatory requirements.
Applicable Regulatory Requirements
Great Britain (England, Scotland, Wales)
-
- The Medical Devices Regulations 2002 (SI 2002 No 618, as amended) (UK MDR (2002))
- The General Product Safety Regulations 2005 (SI 2005 No 1803)*
- Medicines and Medical Devices Act 2021 (MMD Act)
*May apply to consumer products that are also medical devices
Northern Ireland
-
- EU Medical Devices Regulation (2017/745) (MDR)
- EU In Vitro Diagnostic Medical Devices Regulation (2017/746) (IVDR)
- The Medical Devices (Northern Ireland Protocol) Regulations 2021
- The Medical Devices (In Vitro Diagnostic Devices etc.) (Amendment) Regulations 2024
- MMD Act
- The Market Surveillance (Northern Ireland) Regulations 2021
In Great Britain, all medical devices—including IVDs and active implantable medical devices (AIMDs)—are regulated under the UK MDR (2002), which has undergone several revisions following the UK’s exit from the EU on 31 January 2020 (Brexit) and the establishment of the revised EU medical device regulatory framework under the MDR/IVDR. The MHRA is working toward aligning its regulatory framework with the MDR/IVDR; meanwhile, the current framework more closely aligns with the previous EU directives, with certain distinctions, such as UKCA/UKNI marking requirements and the need to assign a UK Responsible Person (UKRP) or UK Importer, as previously discussed in other articles. Among these requirements is mandatory device registration, which applies to all devices, including IVDs, custom-made devices, and systems or procedure packs.
To learn more about MedEnvoy’s UK Importer service, click here.
Key Considerations for Great Britain Medical Device Registration
1. Who Should be Selected as the UKRP?
All medical device manufacturers located outside the UK intending to place their devices on the Great Britain market must assign a UK Responsible Person (UKRP) to serve as their regulatory representative. The UKRP acts on behalf of the manufacturer in a similar role to that of the EU Authorized Representative (EC-REP) assigned by manufacturers located outside the EU and the European Economic Area (EEA) European Free Trade Association (EFTA) states, including registering devices with the MHRA.
Although foreign manufacturers may assign an importer or distributor as their UKRP, it is recommended to select a party independent of the UK distributor due to the regulatory responsibilities associated with the UKRP (such as device registration). This approach reduces the potential regulatory impact of distributor changes, particularly as UKRP information must accompany devices.
2. How are Medical Devices Classified in Great Britain?
Under the UK MDR (2002), the same device classifications established under the former EU Medical Device Directive (MDD) and Active Implantable Medical Device Directive (AIMDD) are used. For IVDs, as under the In Vitro Diagnostic Medical Device Directive (IVDD), classifications follow the same categories established in the EU IVDD—namely, List A, List B, self-testing, and “other.”
3. Can Marketing Authorizations in Other Countries be Leveraged?
Yes. Under amendments to the UK MDR (2002) introduced in June 2023, CE-marked devices can be placed on the UK market according to the following timelines:
-
- The earlier of certificate expiry or 30 June 2028: Devices compliant with the EU Medical Device Directive (MDD) or Active Implantable Medical Device Directive (AIMDD)
-
- The earlier of certificate expiry or 30 June 2030: IVD complaints with the EU In Vitro Diagnostic Medical Device Directive (IVDD)
-
- 30 June 2030: General medical devices, including custom-made devices, complaints with the EU MDR and IVDR
4. What Information is Necessary, and What are the Costs Associated with Great Britain Registration?
For manufacturers who have not yet registered any devices, it is first necessary to register as a manufacturer. If the manufacturer does not have a place of business in the UK, the registration of the manufacturer and its medical devices and IVDs must be conducted by the designated UK Responsible Person.
The following information is required for this registration:

For foreign manufacturers, a letter of designation for the UKRP, which includes a legal contract assigning the UKRP as the exclusive UKRP on behalf of the manufacturer and specifying all mandatory tasks to be undertaken by the UKRP on behalf of the manufacturer.
Once a manufacturer is registered with the MHRA, device registration may be performed, which requires the following information:
-
- Applicable legislation
-
- Device class
-
- Global Medical Device Nomenclature (GMDN) code and term
-
- Basic UDI-DI and UDI-DI(s) (if applicable)
-
- Device name (brand/trade/proprietary name)
-
- Model/version details
-
- Catalogue/reference number
-
- UK Approved Body (or EU Notified Body) (if applicable)
-
- Device attribute information
A complete list of the information necessary for submission, including relevant device attribute information, can be found here.
All device registrations are subject to a £240 statutory fee, with a maximum of 100 devices and a cumulative maximum of 20,000 products per application. Once registered, certain changes must also be notified to the MHRA to maintain up-to-date database information. Below lists the types of changes that are subject to certain statutory fees:
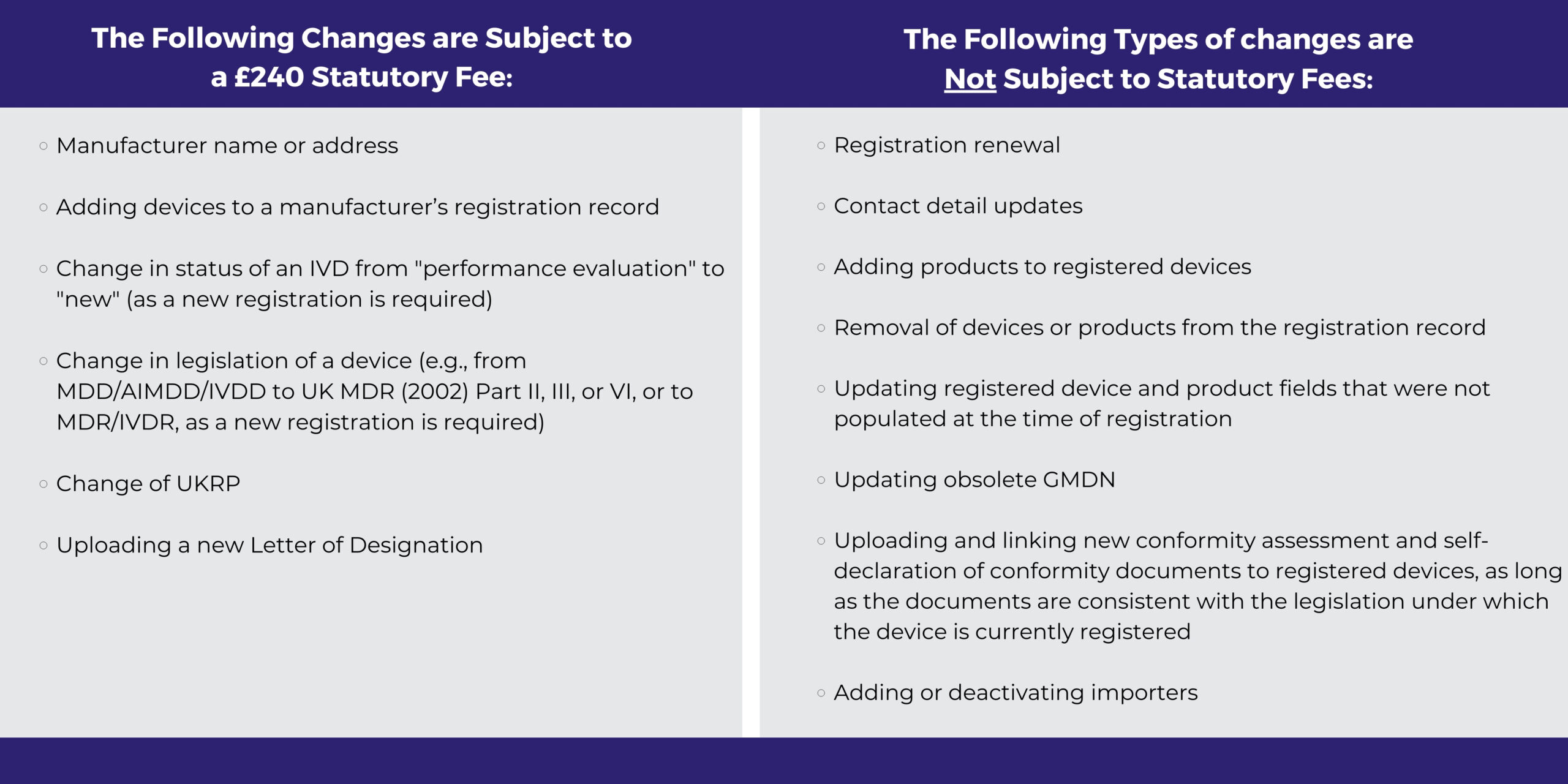
NOTE: On 29 August 2024, the MHRA ran a public consultation on updates to its statutory fees, which closed on 24 October 2024. The MHRA is currently reviewing the feedback obtained; however, manufacturers should prepare for changes to the statutory fees in 2025.
5. What are the Registration Timelines?
Manufacturer registration is relatively fast, typically occurring within 24 hours of submission.
Within five working days, your UK Responsible Person (UKRP) should receive an email confirming the outcome of device registration applications, including whether the MHRA has requested any technical documentation to process the application.
To learn more about MedEnvoy’s UKRP service, click here.
Typically, within the week following registration, details of registered devices will appear on the MHRA Public Access Registration Database (PARD).
It is important for manufacturers to note that device registration with the MHRA does not confer any endorsement, accreditation, certification, or approval by the MHRA. Therefore, manufacturers are not permitted to make any such claims, and it is prohibited to use any MHRA logos in marketing materials, device packaging, instructions for use, or any other documentation.
Learn More About Great Britain Medical Device Registration with MedEnvoy
MedEnvoy’s regulatory experts can assist manufacturers in staying compliant with the latest requirements, including how to obtain conformity assessment and register medical devices in the UK. Furthermore, MedEnvoy has offices in Great Britain and the EU, providing both UK Responsible Person and EU Authorized Representative services. Please reach out if you need assistance by clicking here, and for information about our regulatory experts, click here.


