
Due to the resources necessary to perform a clinical investigation under the MDR, including delays to market entry, the need to perform such investigation is one of the primary considerations for manufacturers during design, development and market selection for commercialization of their devices. Where manufacturers are not applying an Article 61(10) approach to their clinical evaluation, this is of significant concern for manufacturers looking to either maintain or apply the CE mark to their devices. In this article, we cover those devices for which a clinical investigation is mandatory as part of the CE marking process under the MDR and considerations for manufacturers when determining whether such an investigation is necessary for other devices.
When is a Clinical Investigation not Mandatory?
Under Article 61 of the MDR, it is established that clinical investigations must be performed for implantable and Class III devices unless the following circumstances are applicable:
Legacy Devices
The device is a legacy device (i.e. device previously CE marked under the MDD or AIMDD) and for which the MDR clinical evaluation:
-
-
- Is based on sufficient clinical data;
- Complies with the relevant product-specific common specification (CS) for the clinical evaluation of that kind of device, where such a CS is available;
-
Specific Medical Devices with Available Product-Specific Clinical Standards
The device is a suture, staple, dental filling, dental brace, tooth crown, screw, wedge, plate, wire, pin, clip, or connector for which the clinical evaluation is based on:
-
-
- Sufficient clinical data
- Compliance with the relevant product-specific CS, where such a CS is available;
-
Modified Devices by the Same Manufacturer
The device has been designed through modifications of a device already marketed by the same manufacturer with the modified device having been demonstrated to be equivalent to the marketed device by Section 3 of Annex XIV (as endorsed by the Notified Body, after:
-
-
- Checking that the PMCF Plan is appropriate;
- Includes post-market studies to demonstrate device safety and performance),
- The clinical evaluation of the marketed device is sufficient to demonstrate conformity of the modified device with the relevant General Safety and Performance Requirements (GSPR);
-
Equivalency and Clinical Evaluation for Modified Devices from Different Manufacturers
The manufacturer demonstrates equivalency to an already marketed device manufactured by another manufacturer by Section 3 of Annex XIV (as endorsed by the Notified Body, after also checking that the PMCF Plan is appropriate and includes post-market studies to demonstrate device safety and performance), and the clinical evaluation of the marketed device is sufficient to demonstrate conformity of the modified device with the relevant GSPR, so long as (the manufacturer can provide clear evidence that):
-
-
-
- The two manufacturers have a contract in place that explicitly allows the manufacturer of the second device full access to the technical documentation on an ongoing basis, and
- The original clinical evaluation has been performed in compliance with the requirements of the MDR.
-
-
When is it Justified not to Preform Clinical Investigations?
This article also establishes that the EU Commission may exempt other implantable and Class III devices from clinical investigation were justified in the view of well-established technologies, similar to those already exempted devices (i.e. sutures, staples, etc.), or justified in order to protect the health and safety of patients, users or other persons or other aspects of public health.
Therefore, for devices that fulfill the circumstances above, products without an intended medical purpose listed in Annex XVI, and devices in other risk classes (Classes I – IIb (non-implantable)), the manufacturer must demonstrate in the clinical evaluation that there is sufficient clinical data/evidence in the absence of clinical investigation data to justify not performing clinical investigation under the MDR.
What is ‘sufficient clinical data and evidence’?
To determine whether there are sufficient clinical data and evidence for a device, it is important that manufacturers understand the difference between ‘clinical data’ and ‘clinical evidence’ As defined under Article 2 of the MDR:
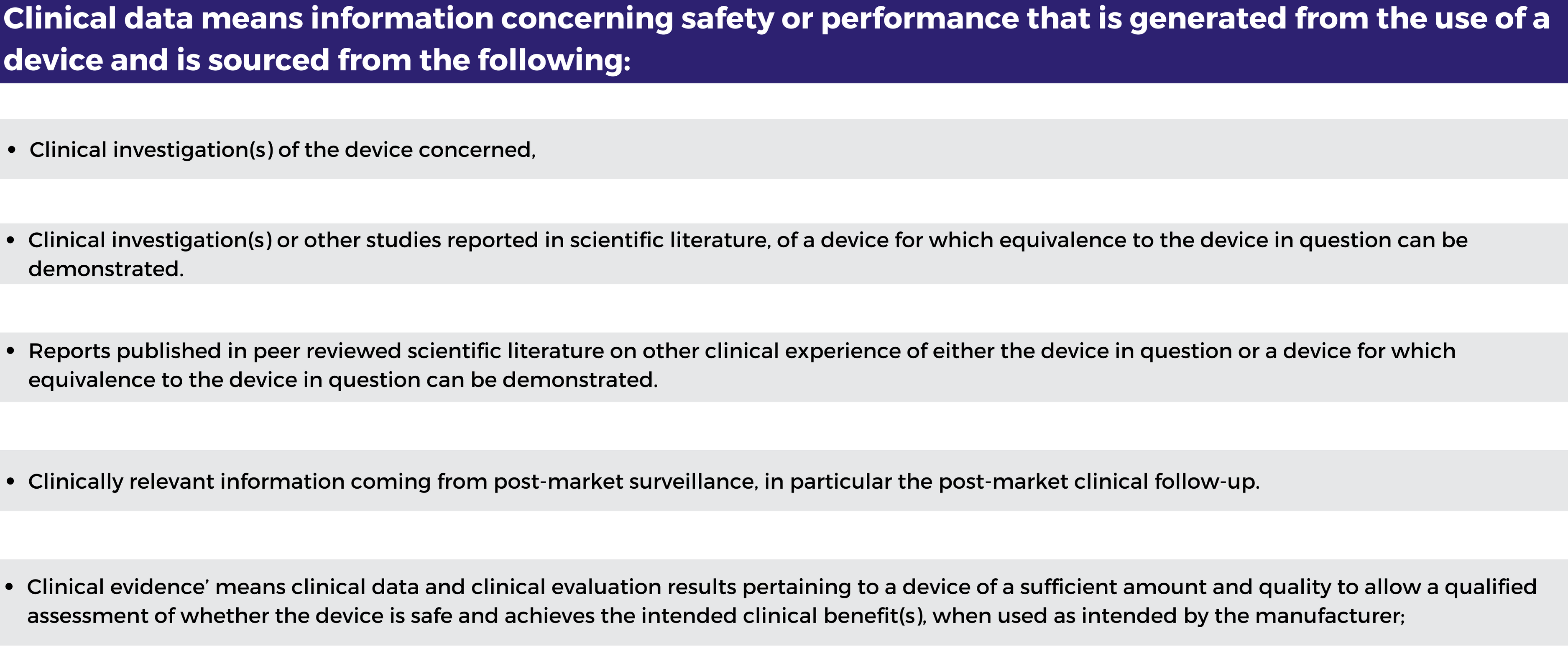
Based upon these definitions, ‘clinical data’ can be understood to be a subset of ‘clinical evidence’ under the MDR. Where manufacturers are not including clinical investigation data under the MDR for their device, the clinical data would comprise a combination of the above types of clinical data of an equivalent device together with post-market surveillance (PMS) / post-market clinical follow-up (data). However, this does not mean that manufacturers must base their clinical evaluations upon equivalency claims, as device novelty and other types of clinical evidence may be used to justify the approach to clinical evaluation.
While MDCG 2020-6 aims to guide clinical data providing sufficient clinical evidence necessary for demonstrating conforming with the relevant GSPR for legacy devices, the guidance may also be considered by non-legacy device manufacturers. In particular, Appendix III of this guidance provides a suggested hierarchy of clinical evidence which ranks twelve different types of clinical data and evidence for manufacturers to consider during clinical evaluation. This includes several types of clinical data/evidence which is not based upon clinical investigations. However, this also requires manufacturers to consider whether their device is ‘well-established technology’, a term not defined under the MDR.
‘Well-established technology’ and Device Novelty
One of the important aspects considered by Notified Bodies in their clinical evaluation assessments is the degree of novelty of the device or whether the device is ‘well-established technology’ as the benefit-risk profile of devices with a low degree of novelty is well characterized across several different types of clinical data, including data in the public domain (e.g. regulatory authority vigilance databases). As noted in MDCG 2020-6, the term ‘well-established technology’ is not restricted only to those devices listed in Article 61(6)(b) of the MDR, but may be considered to include devices that meet ALL of the following criteria:
-
-
- Relatively simple, common, and stable designs with little evolution;
- Their generic device group has well-known safety and has not been associated with safety issues in the past;
- Well-known clinical performance characteristics and their generic device group are standard-of-care devices where there is little evolution in indications and the state of the art;
- A long history in the (EU) market.
-
Therefore, any manufacturer looking to establish an appropriate justification for not performing clinical investigations under the MDR would be well served to demonstrate that their device is a well-established technology and include such criteria and appropriate supporting evidence in their clinical evaluations.
Further consideration should also be given by manufacturers to include a specific section in their clinical evaluations that addresses the degree of device novelty by applying similar criteria to that considered by expert panels through the clinical evaluation consultation procedure established under the MDR. Guidance for this consultation procedure is established in 2020/C 259/02.
This article provides a high-level overview of several considerations for manufacturers in justifying the exclusion of clinical investigation data from their clinical evaluations under the MDR, however, if have any questions regarding clinical evaluation, or require relevant training or consulting services, get in touch.
Learn More About Clinical Investigation Data for CE Marking Under the MDR with MedEnvoy
This article briefly discusses important aspects for manufacturers undergoing device clinical investigations under the MDR. Contact MedEnvoy today to ensure your device meets CE marking standards. MedEnvoy also provides consulting services, learn more about our subject matter experts here.
Related Articles
Related Guidance
We share our expertise because we believe in removing barriers to global commercialization.

Swissmedic Launches 2026 PMS Focus Campaign for Higher-Risk Medical Devices
Swissmedic has issued a formal information letter announcing that it will conduct a focus campaign in 2026 reviewing…

TGA Clarifies Exclusion for Digital Mental Health Software
The Australian Therapeutic Goods Administration (TGA) published guidance on 16 March 2026 clarifying when software products intended…

FDA Reclassifies Two Skin Cancer Diagnostic Devices
The Food and Drug Administration (FDA) issued a final order on March 25, 2026 reclassifying two types of…