
With a projected revenue in 2024 of US $182 billion and an estimated annual growth rate of 5.49% (CAGR 2024-2028), there may be an array of commercial reasons for device manufacturers are debating FDA approval or CE mark market entry due to the US market over the EU-27 market with its projected revenue of US $106.9 billion and estimated annual growth rate of 4.28% (CAGR 2024-2028). This frequently includes the ease or (more often the case) difficulty with which devices may be covered by insurance reimbursement. However, in addition to commercial and logistical considerations, the differences in the regulatory pathways to market are often assessed by organizations when determining market entry priorities.
In this article, we cover several aspects of the respective regulatory pathways in the EU and US that manufacturers are recommended to consider when determining which of these markets to prioritize.
Device Classification in the EU
One of the most immediate considerations for assessing regulatory barriers to entering most major/developed markets is device risk classification, as the controls exerted by regulatory agencies increase correspondingly as device risk class increases. Under the MDR or IVDR, device classification, as determined by the application of the corresponding classification rules, is the primary consideration in determining whether certification through Notified Body (NB) conformity assessment and EU Reference Laboratory (EURL) product testing (in the case of in vitro diagnostic medical devices (IVDs) only) is necessary for the device as follows:
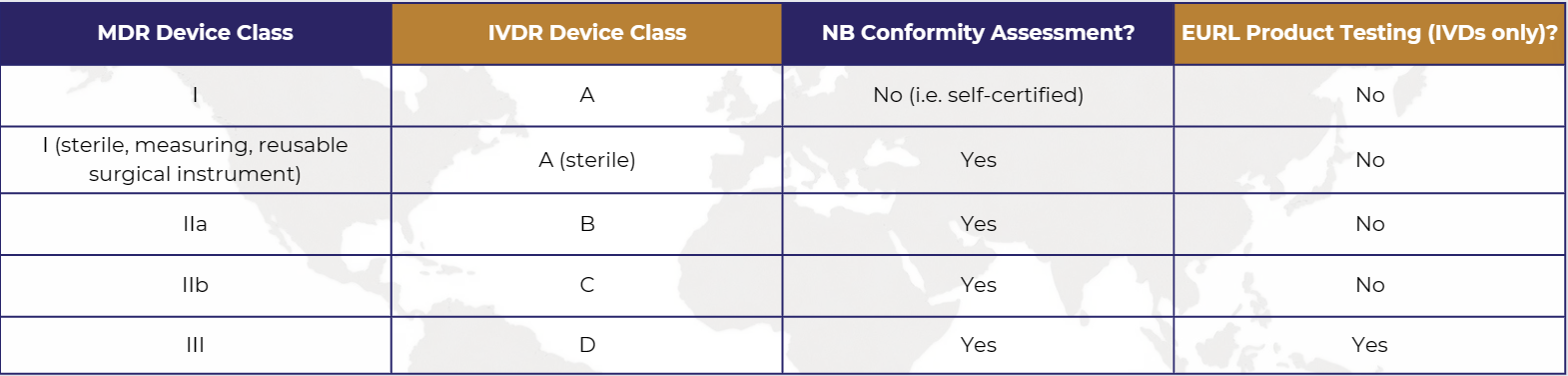
NB conformity assessment will at the very least involve an assessment of the manufacturer’s quality management system (QMS) with the extent of this assessment based on the device classification, the nature of the device, and the conformity assessment procedure applied by the manufacturer, with assessment of product conformity also performed to varying extents (from Technical Documentation File (TDF) review through to product testing) based upon these same considerations.
While manufacturers of self-certified devices avoid these NB assessments, they will still need to establish a QMS based on MDR requirements, however, Full compliance with the EN ISO 13485:2016 standard is not mandatory.
Device Classification in the US
In the case of the US, device classification is not determined through the application of classification rules but rather established by specific device classification regulations, divided into classification panels, which prescribe both the device class and marketing pathway. While general FDA regulatory controls, such as conformity with good manufacturing practice (GMP) requirements and establishment registration/device listing, are applicable across all three risk classes, special controls apply to Class II devices, and pre-market approval (PMA) applies to Class III devices (the highest risk classification). Only manufacturers that have submitted a PMA are subject to establishment inspection by the FDA while any establishment registered with the FDA may be subject to inspection by the FDA at any time. Furthermore, manufacturers of most Class I devices are not obligated to establish design controls as part of their QMS and under some devices are completely exempt from GMP requirements, as described under their respective classification regulation, except record and complaint handling requirements.
Therefore, based upon classification alone, manufacturers can already begin to have an initial understanding of resource requirements for market entry in the EU and US.
Device Novelty in the US
In the case of the US, however, it’s possible that no specific classification regulation currently exists for a device in which case the device is considered novel for that market. The options for US market entry in such circumstances change significantly. While for medium-risk devices, this typically requires a De Novo submission to the FDA, manufacturers may also submit a Section 513(g) Request for Information to obtain the FDA’s view on the classification and regulatory requirements that may apply to their device. In more recent years, the FDA has also made strides in offering other options for what it considers “breakthrough” technologies. Once formal Breakthrfough Device designation has been granted by the FDA, this can help to greatly speed up the pathway for the device, irrespective of the marketing pathway. Where a device is intended to benefit patients in the treatment or diagnosis of a disease or condition that affects or is manifested in not more than 8,000 individuals in the US per year, the Humanitarian Device Exemption (HDE) pathway is available. In general, the US offers several options for novel devices.
Device Novelty in the EU
For the EU, there are fewer options available for manufacturers of novel devices that still need to go through the NB conformity assessment process if they are not subject to self-certification, and in the case of higher-class devices, professional expert consultation is part of the assessment process where a device is the first of its type. Device novelty will also need to be addressed by manufacturers as part of the clinical/performance evaluation process as this is one of the considerations typically expected by NBs to be used to justify the level of clinical evidence presented by the manufacturer. The level of clinical evidence required in the EU will be based on the classification of the device.
In both markets, device novelty is also a significant contributing factor in determining the extent of design and development activities. Companies that have devices with significant novelty will most likely have faster market entry into the EU than the US given that device classification in the EU is not heavily based on equivalent devices but rather on a set of classification rules which are generic in nature. However, if these companies with novelty devices leveraged mechanisms in the US like the Breakthrough Device Designation, this will likely shorten time to market.
Design & Development Considerations in the EU
The foremost concern for manufacturers regarding the impact of device novelty on design and development activities is whether there is a need for clinical investigation (medical devices) or clinical performance studies (IVDs) due to the associated times and costs for the realization of such studies.
In the EU market, where there are low-medium clinical risks related to a device and the manufacturer can identify an “equivalent” device, and to a lesser extent, “similar” devices, data from these devices may be leveraged to varying extents in the device’s clinical/performance evaluation and subsequently used to justify the non-realization of clinical investigation/clinical performance studies. For high-risk devices or novel medium-risk devices, such investigations/studies will most likely be expected by the NB.
Design & Development Considerations in the US
In the US, most devices subject to the 510(k) pathway do not need to present clinical investigational data, although there are notable exceptions, and most IVDs subject to this pathway will need to have clinical performance studies completed.
Further design and development considerations include the need for product certification testing necessary to demonstrate electrical safety and electromagnetic compatibility. However, when assessing other market-specific design inputs, it is important to note that in the case of the EU, there are additional considerations such as:
-
- Localization/Regionalization of device labeling (i.e. translation to comply with Member State national language requirements, including for marketing materials)
- Applicability of other EU legislation such as the EU RoHS Directive, Machinery Directive, Radio Equipment Directive, etc.
One of the main areas in which device manufacturers are struggling with EU design and development requirements is performance evaluation under the IVDR, as this is a new concept to most of them, and current guidance is limited.
In this respect, the US market requirements tend to be more favorable to most device manufacturers.
FDA Approval or CE Mark: Timeframes and Costs
The timeframe is the main consideration for which the US is currently dominates the EU primarily due to the:
-
- 510(k) exempt devices simply require listing under a device establishment with several of these devices also being either GMP exempt or exempt from design control requirements, whereas even self-certified devices in the EU must have a compliant TDF compiled, including clinical/performance evaluation documentation.
- The eSTAR Program, mandatory for all 510(k) submissions since October 2023, has greatly facilitated both the compilation and review process for such submissions.
- There are well-known limits on NB availability and resources for conformity assessment under the MDR and/or IVDR, particularly in the case of the latter, which have resulted in significant delays to manufacturers obtaining CE certification. These limits have resulted in the transition time frames for legacy devices (i.e. devices certified under the previous Directives) being pushed out significantly.
The US is also typically viewed as a more favorable market regarding costs for low-medium risk devices, except the annual establishment for which there is no equivalent under the EU regulatory framework (although the manufacturer does need to pay ongoing certification fees to their NBs where their devices are not self-certified).
Businesses that obtain Small Business Determination (SBD) from the FDA (which must be obtained annually) are eligible for a significant reduction in user fees (e.g. FY 2024 510(k) standard fee = US $21,760; SBD fee = US $5,440). Small businesses with an approved SBD with gross receipts or sales of US $ 30 million or less are also eligible to have the fee waived on their first PMA (Standard fee = US $483,560; SBD fee = US $120,890).
Entry Into Other Markets
Lastly, another consideration when assessing the US FDA or EU CE Marking priorities is whether the CE marking, or US marketing authorization can be leveraged to facilitate entry into other global markets.
Historically, the CE mark has been a greater facilitator for global market entry particularly for those manufacturers considering entry into the UK and Swiss markets, leveraging CE marking continues (for now) to be the easiest means of entry. However, as the FDA is a founding member of the International Medical Device Regulators Forum (IMDRF), and with the progress that has been made over the last several years with the Medical Device Single Audit Program (MDSAP), 510(k) clearance combined with MDSAP certification is beginning to be accepted in other established markets (e.g. Australia). While there are costs associated with obtaining and maintaining MDSAP certification, given the challenges manufacturers have experienced with CE marking under the MDR/IVDR, other regulators have planned proposals to review and possibly implement measures to facilitate market entry for devices with regulatory clearance in established markets (e.g. this topic is on the regulatory agenda for Brazil’s ANVISA in 2024-2025). Such measures would greatly swing the balance towards US marketing authorization combined with MDSAP certification.
FDA Approval or CE Mark: What to Prioritize
While device characteristics, particularly classification and novelty, will continue to be the main regulatory drivers in prioritizing FDA approval or CE mark market entry, the information presented in this article suggests that for many manufacturers the US market has the lowest barriers to entry, with the potential for this gap to grow further over the next few years.
This article provides an overview of regulatory considerations when assessing EU vs US FDA device market entry. If you have any questions regarding this topic or require relevant consulting support or regulatory representation in either of these markets, get in touch.
Related resources:



Related Guidance
We share our expertise because we believe in removing barriers to global commercialization.

COFEPRIS Finalizes Medical Device Labeling Standard
On May 19, COFEPRIS published an updated medical device labeling standard, NOM-137-SSA1-2025, in the official Diario Oficial de la Federación (DOF). The update replaces…

Swissmedic Launches 2026 PMS Focus Campaign for Higher-Risk Medical Devices
Swissmedic has issued a formal information letter announcing that it will conduct a focus campaign in 2026 reviewing…

TGA Clarifies Exclusion for Digital Mental Health Software
The Australian Therapeutic Goods Administration (TGA) published guidance on 16 March 2026 clarifying when software products intended…