
The Therapeutic Goods Administration (TGA) Unique Device Identification (UDI) system has been in development for several years with several rounds of public consultation and opportunities for the industry to provide feedback and engage with the regulator. In 2024 the UDI regulatory framework and compliance dates are expected to begin. The TGA’s UDI Hub is the central resource for manufacturers to prepare for the transition with this article providing an overview of the resources available and the TGA UDI requirements.
The TGA UDI Regulatory Framework
While the UDI regulations, implementation timeframes, and compliance dates for the TGA’s UDI regulatory framework have yet to be published, it has already published information on the expectations for the system based on the feedback already received. The TGA has also indicated that UDI requirements, including labeling and provision of UDI data, will be part of the Essential Principles established under Schedule 1 of the Therapeutic Goods (Medical Devices) Regulations 2002.
Alignment with Essential Principles
One of the key aims of the TGA has been to minimize the burden of yet another UDI system on manufacturers who have already had to comply with such requirements in other jurisdictions. Therefore, the framework has been aligned with current IMDRF guidance, recognizes UDI issuing agencies that are already internationally accepted (GS1, HIBCC, ICCBBA), and allows for the acceptance of EU and US UDI-compliant labeling with minimization of Australian-only requirements.
There is no equivalent to the EU “Basic UDI-DI” under the Australian framework but the UDI will be comprised of two parts, the UDI-Device Identifier (UDI-DI) (e.g. GS1 Global Trade Item Number (GTIN)) which identifies the model of the device and UDI-Product Identifier (UDI-PI) which identifies the production-specific information such as batch/lot number or expiry date.
Australian TGA Sponsors will be responsible for ensuring that, where applicable:
-
-
- Any UDIs on labels on the device and all higher levels of packaging can be both human and machine-readable and, where required, the UDI is directly marked on the device.
-
-
-
- UDI and related data is submitted to the Australian UDI Database (AusUDID) and correctly linked to the corresponding Australian Register of Therapeutic Goods (ARTG) entry for the device in question
- Ensuring that UDI data is maintained up-to-date while supplying the device
-
Compliance Phases and Exemptions
As with other jurisdictions, mandatory compliance with UDI registration and labeling requirements will be progressively phased in based on device classification, beginning with high-risk/implantable devices for which compliance is expected to begin at a minimum of 12 months from the date the regulation comes into effect.
UDI requirements will be mandatory for the following devices:
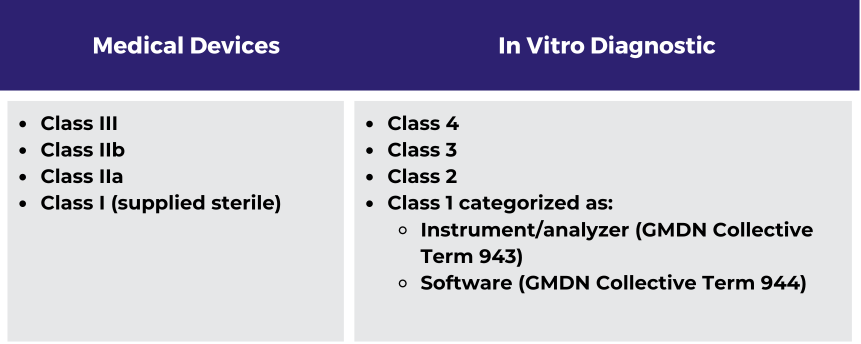
Phased Implementations and UDI Requirements
While manufacturers can choose to voluntarily comply with the UDI requirements at any time once the regulations come into effect there are several devices exempt from meeting UDI requirements, such as:
-
-
- Export only devices / IVDs
- Custom made devices
- Patient-matched devices with a volume of 5 units or less supplied each financial year (with the Australian financial year beginning on July 1)
- Devices exempt under Special Access Scheme (SAS) or Authorized Prescriber Scheme (APS)
- Class 1, 2, and 3 in-house IVDs
-
Transition Periods and Direct Marking
For Class III and IIb devices manufactured prior to their mandatory compliance dates, manufacturers will have a 3-year transition period to ensure that they comply with UDI labeling and data provision requirements whereas all other classes of devices manufactured prior to their labeling compliance dates are exempt from complying with these requirements during their lifetime (this exemption also applies to devices requiring direct marking). However, this exemption does not apply where existing devices are relabeled or remanufactured after their original supply date and after the mandatory compliance dates.
Transition Periods for Class III and IIb Devices
Direct marking requirements will be applicable to devices reprocessed between use on subsequent patients with manufacturers having to determine the application of machine-readable or human-readable carriers based upon the expected end-use when there are space limitations on the device. Furthermore, direct marking exemptions are applicable for:
-
-
- Devices reprocessed between uses on the same patient
-
-
-
- Implantable devices
- Where direct marking would interfere with device safety, performance, or effectiveness
- It is not technologically feasible to directly mark the device
-
UDI Documentation and Reporting
Lastly, where a UDI is available, and once the regulations are in force, the UDI will be included on Patient Implant Cards (PICs) and notifications submitted to the TGA including adverse events, incident reports, and recalls.
Resources for Industry
The TGA has proactively taken a highly consultative approach in preparing for the transition to UDI, including the rollout of a pilot version of the AusUDID referred to as a ‘sandpit’ that allowed players from across all areas of the device industry to trial the database and provide feedback. While AusUDID will be launched once the UDI regulations take effect, the pre-production version will continue to be made available by the TGA so that sponsors and manufacturers may test its functionality and their preferred data submission methods prior to actual submission.
Continuing its highly interactive approach to the development of the UDI regulatory framework, the TGA has established its UDI Hub and set up a dedicated TGA UDI team that can be contacted with any queries regarding the UDI system (UDI@health.gov.au).
In addition to being able to subscribe for news and updates on the UDI system, the two primary resources for manufacturers are:
-
-
- Information for sponsors and manufacturers – This resource offers an overview of UDI requirements, guiding stakeholders through the compliance process.
- Resources and technical documents – Currently containing draft versions of technical documents, final versions will be made available in 2024, this page also includes access to the public consultations previously held by the TGA and relevant webinars that it has administered.
-
Learn More About the TGA UDI Regulatory Framework with MedEnvoy
In anticipation of the imminent release of its UDI regulations, this article provides an overview of the TGA UDI regulatory framework and TGA UDI Hub. If you have any questions regarding Australian medical device regulatory requirements or require an Australian Sponsor, get in touch.
Related resources:



Related Guidance
We share our expertise because we believe in removing barriers to global commercialization.

Swissmedic Launches 2026 PMS Focus Campaign for Higher-Risk Medical Devices
Swissmedic has issued a formal information letter announcing that it will conduct a focus campaign in 2026 reviewing…

TGA Clarifies Exclusion for Digital Mental Health Software
The Australian Therapeutic Goods Administration (TGA) published guidance on 16 March 2026 clarifying when software products intended…

FDA Reclassifies Two Skin Cancer Diagnostic Devices
The Food and Drug Administration (FDA) issued a final order on March 25, 2026 reclassifying two types of…